IgPhyML lineage tree analysis¶
This is the instruction guide for using the new, repertoire-wide version of IgPhyML. A description of HLP17, the model underlying IgPhyML, can be found here:
Hoehn, K. B., Lunter, G., & Pybus, O. G. (2017). A phylogenetic codon substitution model for antibody lineages. Genetics, 206(1), 417-427. doi:http://dx.doi.org/10.1534/genetics.116.196303
Warning
The new repertoire-wide version of IgPhyML isn’t officially released yet, so please let us know if you’re planning to publish anything using it. A lot of the features are still in active development, so feedback, especially with input/output format and problems encountered, would be greatly appreciated.
Also, if you’re analysing single lineages, all of the old features
still work, so check out the old manual for that
(see docs/IgPhyML_Manual.pdf in the IgPhyML source bundle).
Installation¶
Download and install the new IgPhyML:
git clone https://bitbucket.org/kbhoehn/igphyml
cd igphyml
Linux¶
On Linux operating systems, you can usually just run:
./make_phyml_blas_omp
If these commands dont work, you may need to install BLAS and LAPACK,
which provide libraries for better doing matrix exponentiation
operations. In Ubuntu Linux, these are provided in the packages
libblas-dev and liblapack-dev. Other distros probably have
similar package names. If you still can’t get this to work, IgPhyML
can be compiled with OpenMP but without BLAS, though this may negatively
affect performance:
./make_phyml_omp
or without OpenMP as well (though this will slow analysis considerably):
./make_phyml
Once everything is compiled, just add the igphyml/src directory to your
PATH variable, and IgPhyML should work. Importantly, some directory
files are hardcoded in at compilation, so re-compile IgPhyML if you move
the installation directory. Alternatively, you can set the IGPHYML_PATH
environment variable to the location of the igphyml/src/motifs folder for
the same effect.
Mac OS X¶
Installation on Mac OS X is trickier, but possible. The primary issue
is gaining OpenMP support, and installing some GNU command line tools.
The best way is to just install the latest version of llvm
available through homebrew, as well as autoconf and
automake. To do these you’ll need to install
homebrew. If it’s already installed be
sure it’s at the latest version (brew update). You may need to install
Xcode as well. Next, install autoconf, automake, and llvm:
brew install autoconf
brew install automake
brew install llvm
Specify the llvm version of clang in Makefile.am and
src/Makefile.am by adding the line CC=<path to llvm clang>
to the beginning of both files. You will also need to add
MACOMP=<path to omp.h> and MACLLVM=<path to llvm lib> to
src/Makefile.am. For instance, if you’ve install llvm 3.9.1
via homebrew, you will likely need to add the line
CC=/usr/local/Cellar/llvm/3.9.1/bin/clang
to Makefile.am and the lines:
CC=/usr/local/Cellar/llvm/3.9.1/bin/clang
MACOMP=/usr/local/Cellar/llvm/3.9.1/lib/clang/3.9.1/include/omp.h
MACLLVM=/usr/local/Cellar/llvm/3.9.1/lib
to src/Makefile.am.
Your specific path may look different, but you can check locations
of these files and folders by looking around in
/usr/local/Cellar/llvm/. The directory structure should be
similar. Run ./make_blas_phyml_omp, or other versions, as desired, and add
the src folder to your PATH variable.
On some versions of OS X it may be necessary to install XCode command line tools using:
xcode-select --install
cd /Library/Developer/CommandLineTools/Packages/
open macOS_SDK_headers_for_macOS_<OS X version>.pkg
Quick start¶
These commands should work as a first pass on many reasonably sized datasets, but if you really want to understand what’s going on or make sure what you’re doing makes sense, please check out the rest of the manual.
Convert Change-O files into IgPhyML inputs
Move to the examples subfolder and run:
BuildTrees.py -d example.tab --outname ex --log ex.log --collapse \
--sample 3000 --minseq 2
Build lineage trees using the GY94 model (fast, doesn’t correct for hotspots):
igphyml --repfile ex_lineages.tsv -m GY
Build lineage trees using the HLP19 model (slower, corrects for WRC/GYW hotspots):
igphyml --repfile ex_lineages.tsv -m HLP
Both of these can be parallelized by adding
--threads <thread count> option. Trees files are listed as
ex/<clone id>.fasta_igphyml_tree.txt, and can be viewed with most
tree viewers (I recommend
FigTree). Parameter
estimates are in ex_lineages.tsv_igphyml_stats.txt. Subsampling using
the --sample option in BuildTrees isn’t strictly necessary, but
IgPhyML will run slowly when applied to large datasets.
Processing Change-O data sets¶
The process begins with a Change-O formatted data file, in
which each sequence has been clustered into a clonal group,
which has subsequently had its unmutated V and J sequence predicted.
The following column names are required in the input file: fields: SEQUENCE_ID,
SEQUENCE_INPUT, SEQUENCE_IMGT, GERMLINE_IMGT_D_MASK,
V_CALL, J_CALL, and CLONE. FUNCTIONAL is recommended.
Use BuildTrees to break this file into separate sequence alignment files that can be used with IgPhyML. This program will:
- Filter out nonfunctional sequences.
- Mask codons split by insertions.
- Separate clonal groups into separate alignment files (aligned by IMGT site) and information files
- Create the repertoire files for this dataset.
Create IgPhyML input files from examples/example.tab:
cd examples
BuildTrees.py -d example.tab --outname ex --log ex.log --collapse
This will create the directory ex and the file
ex_lineages.tsv. Each ex/<clone ID>.fasta contains the IMGT
mutliple sequence alignemt for a particular clone, and each
ex/<clone ID>.part.txt file contains information about V and J
germline assignments, as well as IMGT unique numbering for each site.
The file ex.log will contain information about whether or not each
sequence was included in the analysis. The file ex_lineages.tsv is
the direct input to IgPhyML. Each line represents a clone and shows
the multiple sequence alignment, starting tree topology (N if
ignored), germline sequence ID in alignment file, and partition file
(N if ignored). These repertoire files start with the number of
lineages in the repertoire, and lineages are arranged from most to
least number of sequences. Here, the --collapse flag is used to
collapse identical sequences. This is highly recommended because
identical sequences slow down calculations without actually affecting
likelihood values in IgPhyML.
Subsampling Change-O datasets
IgPhyML runs slowly with more than a few thousand sequences. You can
subsample your dataset using the --sample and --minseq options,
which will subsample your dataset to the specified depth and then remove
all clones below the specified size cutoff:
BuildTrees.py -d example.tab --outname ex --log ex.log --collapse --sample 5 --minseq 2
Removing the CDR3 region
If you plan to analyze model parameters to study things such as SHM and
selection (see :ref: below <igphyml-parameters>), it’s important to remove
the CDR3 region to avoid known model biases in estimating 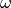 . To
do this, use
. To
do this, use --ncdr3:
BuildTrees.py -d example.tab --outname ex --log ex.log --collapse --ncdr3
Note
IgPhyML requires at least three sequences in a lineage, so in the case that there is only one observed sequence within a clone, that sequence is duplicated. This will not affect the likelihood calculation because these sequences will have a branch length of zero, but it will affect metrics that take sequence frequency into account.
Building B cell lineage trees¶
Before doing any further analysis, I strongly recommend estimating intitial tree topologies using the GY94 model. This can improve runtime for later analysis:
igphyml --repfile ex_lineages.tsv -m GY --outrep ex_lineages.GY.tsv --run_id GY
Here, the data files are specifed with --repfile. Topologies are
searched using NNI moves. To do a more thorough topology search, use
-s SPR. The flag --outrep will create a repertoire file that is
identical to the file specified in --repfile but with the resulting
GY94 topologies specified for each lineage. We can view the ML
parameter estimates for the GY94 fit in
ex_lineages.tsv_igphyml_stats_GY.txt, and the tree topologies for
each clone individual lineage in
ex/<clone id>.fasta_igphyml_tree_GY.txt. I recommend using
FigTree to visualize
topologies.
To estimate ML tree topologies using the HLP19 model wth a GY94 starting topology, use:
igphyml --repfile ex_lineages.GY.tsv -m HLP --run_id HLP --threads 2
This will estimate a single ,  , set of
codon frequencies (
, set of
codon frequencies (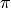 ), and WRC/GYW mutability across the
entire repertoire, and search for topologies using NNI moves. You can
see parameter estimates in
), and WRC/GYW mutability across the
entire repertoire, and search for topologies using NNI moves. You can
see parameter estimates in
ex_lineages.GY.tsv_igphyml_stats_HLP.txt, and trees in
ex/<clone id>.fasta_igphyml_tree_HLP.txt. This command will also
parallelize the calculation across 2 threads using the --threads
flag.
Phylogenetic model parameter analysis¶
The HLP19 model is the heart of IgPhyML and adjusts for features of affinity maturation that violate the assumptions of most other phylogenetic models. It uses four sets of parameters to characterize the types of mutations the occurred over a lineage’s development, and to help build the tree.
: Also called dN/dS, or the ratio of nonsynonymous
(amino acid replacement) and synonymous (silent) mutation rates. This
parameter generally relates to clonal selection, with totally neutral
amino acid evolution having an 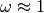 , negative
selection indicated by
, negative
selection indicated by 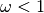 and diversifying selection
indicated by
and diversifying selection
indicated by  . Generally, we find a lower
for FWRs than CDRs, presumably because FWRs are more structurally
constrained.
. Generally, we find a lower
for FWRs than CDRs, presumably because FWRs are more structurally
constrained.
: Ratio of transitions (within purines/pyrimidines) to
transversions (between purines/pyrimidines). For normal somatic
hypermutation this ratio is usually 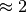 .
.
Motif mutability (e.g. 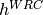 ): Mutability parameters for
specified hot- and coldspot motifs. These estimates are equivalent to
the fold-change in mutability for that motif compared to regular
motifs, minus one. So,
): Mutability parameters for
specified hot- and coldspot motifs. These estimates are equivalent to
the fold-change in mutability for that motif compared to regular
motifs, minus one. So, 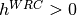 indicates at hotspot,
indicates at hotspot,
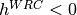 indicates a coldspot, and
indicates a coldspot, and 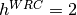 indicates a 3x increase in WRC substitution rate. The HLP19 model
by default estimates six motif mutability parameters: four hotspots
(WRC, GYW, WA, and TW) and two coldspots (SYC and GRS).
indicates a 3x increase in WRC substitution rate. The HLP19 model
by default estimates six motif mutability parameters: four hotspots
(WRC, GYW, WA, and TW) and two coldspots (SYC and GRS).
Substitution models are specified using the -t for
(transition/transverion rate), --omega for
(nonsynonymous/synonymous mutation rate), and --motifs and
--hotness for specifying the motif mutability models. The default
for all of these is to estimate shared parameter values across all
lineages, which is also specified by e.
Due to default parameter settings, the following two commands are equivalent:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr
igphyml --repfile ex_lineages.GY.tsv -m HLP -t e --omega e,e \
--motifs WRC_2:0,GYW_0:1,WA_1:2,TW_0:3,SYC_2:4,GRS_0:5 \
--hotness e,e,e,e,e,e -o lr
In both cases parameter estimates are recorded in
ex_lineages.GY.tsv_igphyml_stats.txt. Note that here we use
-o lr, which will keep tree topologies the same and only estimate
branch lengths and substitution parameters. This will keep topologies
the same as the GY94, but will estimate substitution parameters much
more quickly.
Confidence interval estimation
It is possible to estimate 95% confidence intervals for any of these
parameters by adding a ‘c’ to the parameter specification. For example,
to estimate a 95% confidence interval for in the CDRs
but not the FWRs, use:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr --omega e,ce
To estimate a 95% confidence interval for in the FWRs
but not the CDRs, use:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr --omega ce,e
Any combination of confidence interval specifications can be used
for the above parameter options. For motif mutability, each value
in the --hotness option corresponds to the index specified in
the --motifs option. To estimate confidence intervals for GYW
mutability, use:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr \
--hotness e,ce,e,e,e,e
which is equivalent to:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr \
--motifs WRC_2:0,GYW_0:1,WA_1:2,TW_0:3,SYC_2:4,GRS_0:5 \
--hotness e,ce,e,e,e,e
You can also alter constrain motif to have the same mutabilities
by altering the indexes after the ‘:’ in the --motifs option.
For example, to estimate 95% confidence intervals on a model in
which WRC/GYW, WA/TW, and SYC/GRS motifs are respectively constrained
to have the same mutabilities, use:
igphyml --repfile ex_lineages.GY.tsv -m HLP -o lr \
--motifs WRC_2:0,GYW_0:0,WA_1:1,TW_0:1,SYC_2:2,GRS_0:2 \
--hotness ce,ce,ce
You can find further explanation of the different options in the commandline help, including controlling output directories and file names.
Optimizing performance¶
IgPhyML is a computationally intensive program. There are some ways to make calculations more practical, however:
Data subsampling: IgPhyML runs slowly with more than a few thousand sequences. You can
subsample your dataset using the --sample and --minseq options in
BuildTrees, which will subsample your dataset to the specified depth and
then remove all clones below the specified size cutoff (see Subsampling
Change-O datasets).
Parallelizing computations: It is possible to parallelize likelihood
calculations using the --threads option. Currently, calculations
are parallelized by tree, so there is no point in using more threads
than you have lineages in your repertoire file.
GY94 starting topologies: Calculations are much faster under the GY94
model, so it is usually better to do initial topology
searching under the GY94 model,
and then use those trees as starting topologies for HLP19. You can also
keep these topologies the same during HLP19 parameter estimation (-o lr)
for an even greater speedup, though, obviously, this will not result in a
change in topology from GY94.
Enforcing minimum lineage size: Many repertoires often contain huge
numbers of small lineages that can make computations impractical. To
limit the size of lineages being analyzed, specify a cutoff with
--minseq when running BuildTrees. IgPhyML has a --minseq option
with the same functionality, but this option includes the predicted germline
sequence and duplicated sequences in singleton clones. Because of this,
it is recommended to do --minseq filtering at the BuildTrees step.