Clonal clustering methods
The DefineClones.py tool provides multiple different approaches to assigning Ig sequences into clonal groups.
Clustering by V-gene, J-gene and junction length
All methods provided by DefineClones.py first partition sequences based on
common IGHV gene, IGHJ gene, and junction region length. These groups are then
further subdivided into clonally related groups based on the following distance
metrics on the junction region. The specified distance metric
(--model) is then
used to perform hierarchical clustering under the specified linkage
(--link) clustering. Clonal groups are
defined by trimming the resulting dendrogram at the specified threshold
(--dist).
Amino acid model
The aa distance model is the Hamming distance
between junction amino acid sequences.
Hamming distance model
The ham distance model is the Hamming
distance between junction nucleotide sequences.
Human and mouse 1-mer models
The hh_s1f and
mk_rs5nf distance models are single
nucleotide distance matrices derived from averaging and symmetrizing the human 5-mer
targeting model in [YVanderHeidenU+13] and the mouse 5-mer targeting model in
[CDiNiroVanderHeiden+16]. The are broadly similar to a transition/transversion model.
Human 1-mer substitution matrix:
Nucleotide |
A |
C |
G |
T |
N |
|---|---|---|---|---|---|
A |
0 |
1.21 |
0.64 |
1.16 |
0 |
C |
1.21 |
0 |
1.16 |
0.64 |
0 |
G |
0.64 |
1.16 |
0 |
1.21 |
0 |
T |
1.16 |
0.64 |
1.21 |
0 |
0 |
N |
0 |
0 |
0 |
0 |
0 |
Mouse 1-mer substitution matrix:
Nucleotide |
A |
C |
G |
T |
N |
|---|---|---|---|---|---|
A |
0 |
1.51 |
0.32 |
1.17 |
0 |
C |
1.51 |
0 |
1.17 |
0.32 |
0 |
G |
0.32 |
1.17 |
0 |
1.51 |
0 |
T |
1.17 |
0.32 |
1.51 |
0 |
0 |
N |
0 |
0 |
0 |
0 |
0 |
Human and mouse 5-mer models
The hh_s5f and
mk_rs5nf distance models are based on
the human 5-mer targeting model in [YVanderHeidenU+13] and mouse 5-mer
argeting models in [CDiNiroVanderHeiden+16], respectively. The targeting
matrix 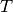 has 5-mers across the columns and the nucleotide to
which the center base of the 5-mer mutates as the rows. The value for a
given nucleotide, 5-mer pair
has 5-mers across the columns and the nucleotide to
which the center base of the 5-mer mutates as the rows. The value for a
given nucleotide, 5-mer pair ![T[i,j]](../_images/math/b40a549ad41eda2fc5b0b704307deba40dd1de03.png) is the product of the
likelihood of that 5-mer to be mutated
is the product of the
likelihood of that 5-mer to be mutated 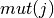 and the
likelihood of the center base mutating to the given nucleotide
and the
likelihood of the center base mutating to the given nucleotide
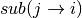 . This matrix of probabilities is converted
into a distance matrix
. This matrix of probabilities is converted
into a distance matrix 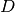 via the following steps:
via the following steps:
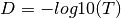
- is then divided by the mean of values in
All distances in
that are infinite (probability of zero),
distances on the diagonal (no change), and NA distances are set to 0.
Since the distance matrix is not symmetric, the
--sym argument
can be specified to calculate either the average (avg) or minimum (min)
of 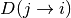 and
and 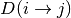 .
The distances defined by for each nucleotide difference are
summed for all 5-mers in the junction to yield the distance between the
two junction sequences.
.
The distances defined by for each nucleotide difference are
summed for all 5-mers in the junction to yield the distance between the
two junction sequences.
Ang Cui, Roberto Di Niro, Jason A. Vander Heiden, Adrian W. Briggs, Kris Adams, Tamara Gilbert, Kevin C. O'Connor, Francois Vigneault, Mark J. Shlomchik, and Steven H. Kleinstein. A Model of Somatic Hypermutation Targeting in Mice Based on High-Throughput Ig Sequencing Data. The Journal of Immunology, 197(9):3566–3574, nov 2016. URL: http://www.jimmunol.org/content/197/9/3566.abstract http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.1502263, doi:10.4049/jimmunol.1502263.
Gur Yaari, Jason A Vander Heiden, Mohamed Uduman, Daniel Gadala-Maria, Namita Gupta, Joel N H Stern, Kevin C O'Connor, David A Hafler, Uri Laserson, Francois Vigneault, and Steven H Kleinstein. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Frontiers in immunology, 4:358, January 2013. URL: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3828525\&tool=pmcentrez\&rendertype=abstract, doi:10.3389/fimmu.2013.00358.